尊龙app下载 什么是化学键? 共价 / 离子 / 金属 / 配位键各异梳理与电催化 “键强 — 活性” 关联中枢
发布日期:2026-03-14 12:12 点击次数:162
说明:本文采算科技从化学键的骨子开赴,梳理共价、离子、金属与配位键的各异,强调电催化中“键强—活性”关联。以DOS/PDOS与d带中心形容吸附轨说念重排,用COHP/COOP与ICOHP定量键强,并辅以Bader电荷与差分电荷密度追踪电子滚动。
围绕Pt–CO抗中毒、RuO2高熵掺杂触发OPM、单原子在N掺石墨烯上的锚定,以及ARSC等数据驱动神态符,展示“调键即调活性”的策动范式,劝诱机阐明析与材料优化。
什么是化学键?
从骨子上说,化学键着手于原子间电子的从头散布,包括共价键、离子键和金属键等。在催化剂与反应物的相互作用中,时常波及配位键和键的断裂/变成历程。阐明这些键的强度与本性,对于发达反应机制和性能至关蹙迫。
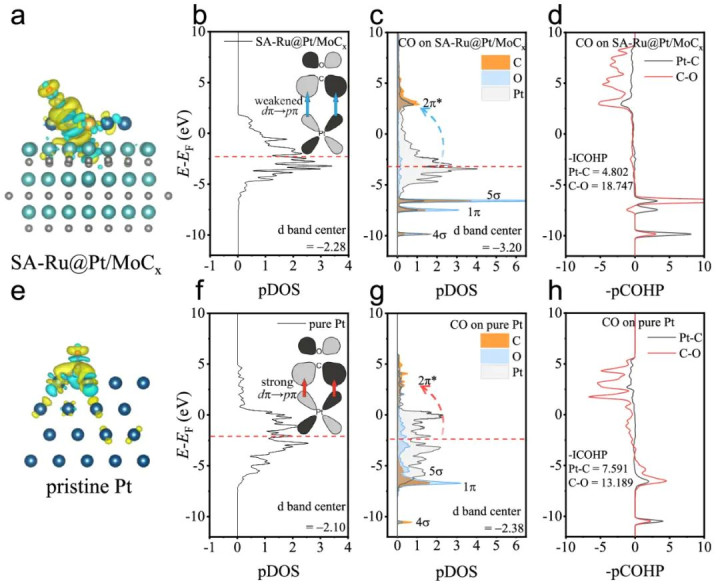
DOI:10.1038/s41467-024-51562-9
通过比较催化剂在反应前后的DOS变化,不错了解吸附变成化学键时电子态的重排。举例,吸附物轨说念在催化剂d带中的投影峰位置和强度反馈轨说念重复程度,从而指令键合强度。
DOS还用于谋略d带中心等参数。d带中心是表征过渡金属名义价层d电子能级位置的一个简化主见,宽泛用加权平均能量示意。d带中心接近费米能级意味着d电子能级上移,提供更多填充反键轨说念的智商,从而时常加强对吸附物的键合营用;反之d带中心下移则削弱吸附。
经典的d带模子还是被诠释对阐明过渡金属名义的键变成和反应性趋势相称有用。可是,浅陋的d带中心并非适用于总计体系,连年来一些责任提议探讨d带时局(如宽度和填充度)的更始神态符,以更精确地预测吸附性质。
晶体轨说念哈密顿布居(COHP)或晶体轨说念重复布居(COOP)方法,将化学键的态密度按成键(沉静化)和反键(不沉静)孝顺拆分。集成的COHP值(ICOHP)可定量推敲两个原子间键合营用强度(数值王人备值越大示意键越强)。
举例,在Pt催化剂上吸附CO的照看中,对Pt–C键的-ICOHP值进行比较发现:调控后的催化剂Pt–C键ICOHP显耀裁减(键弱化),而CO里面C–O键ICOHP增大(键加强),标明Pt对CO的吸附减轻、CO自身键更郑重。这种成键/反键态分析有助于阐明催化剂若何通过电子结构调理影响吸附键的强弱。
基于电子密度分割的方法,将每个原子占有的电子数进行诀别,从而得到原子部分电荷。Bader电荷提供了键合历程中电荷滚动的定量神态:正的Bader电荷增量意味着原子失电子、成为电子穷乏;负的则相悖。通过比较反应前后的Bader电荷,可判断哪种原子/物种在变成化学键时获取或失去电子。
举例,在高密度单原子催化剂中,跟着单原子负载密度增多,相邻位点之间产生电荷从头散布,Co原子的Bader净电荷变得更低(意味着Co失去电子变得更正)而d带中心上移,这种电子结构变化被诠释成心于O2等物种的活化。
通过将吸附体系的总电子密度减去各组成部分(催化剂名义+单独吸附物)的电子密度,不错得到吸附引起的电荷重排三维散布。
CDD宽泛用等值面图显露:其中电子累积区域(一般用黄色示意)和电子耗散区域(青色)直不雅揭示化学键变成时电子从何处滚动到了何处。举例,电催化剂名义吸附小分子后,差分电荷密度图不错显露金属与吸附种之间变成键时电子云在两者之间聚合,以及金属原子失去一定电子密度等。
这种图像化妙技有助于阐明供电子/受电子(如σ给以和π反馈)历程是否发生,从而与经典键合模子相对应。
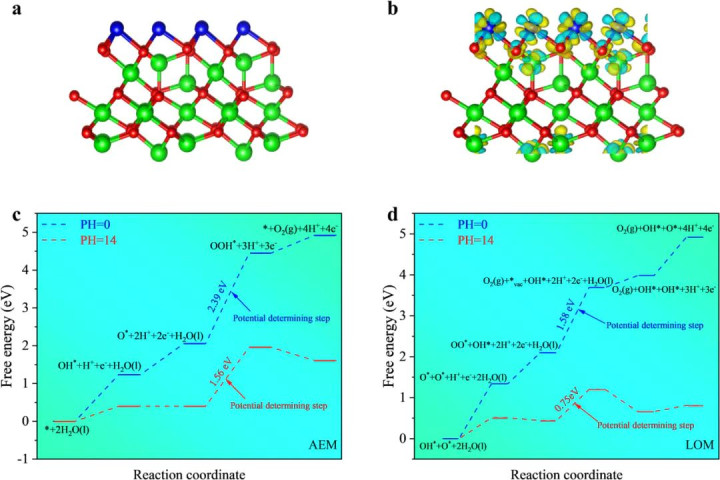
DOI:10.1016/j.cej.2025.165083
化学键有什么用?
调控催化活性
催化剂活性的上下与反应物/中间体在其名义的化学键强度密切有关。若是键过强,吸附种难以脱附或中间体过于沉静会扼制反应程度;若键过弱,又不及以有用吸附活化反应物。
因此,通过调控催化剂–吸附物键强来优化催化活性是电催化照看的中枢想路之一。在过渡金属电催化剂中,d电子在化学键中的作用尤为要津——这恰是d带中神态论广受关爱的原因。
Nørskov等配置的d带模子指出,金属d带电子结构与其吸附键强呈系统有关,已成为解释过渡金属催化剂名义键合和反应活性各异的基础表面。
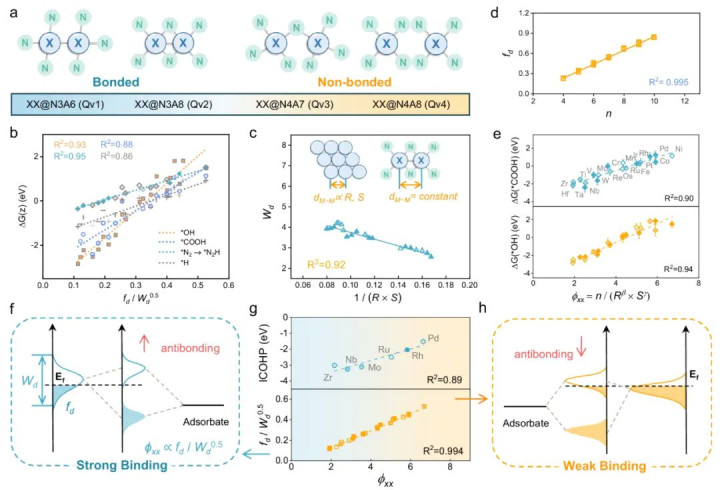
DOI:10.1038/s41467-024-52519-8
连年来多数顶刊照看标明,通过合金化、结构策动等妙技调控金属d带占据和能级位置,不错有用改变催化剂的键合智商,从而晋升特定反应的活性。举例,CO分子在Pt名义的中毒效应家喻户晓,为提高Pt催化剂在富CO环境下的氢气氧化反应活性,要津在于削弱Pt–CO键。
证据Blyholder经典模子,CO吸附在Pt上的键合着手于CO的5σ轨说念向金属d轨说念提供电子以及金属d轨说念向CO的2π反键轨说念反馈电子。
因此,只须大约适合裁减Pt d轨说念对CO 2π的回馈,就能减轻Pt–CO键强度,从而缓解CO中毒。具体罢了方法上,照看者通过多种策略来裁减Pt的d带中心:举例将Pt与过渡金属合金化、构筑核壳结构、或利用氧化物载体使Pt产生电荷滚动,这些都会使Pt d轨说念能级着落,减少d电子占据,从而削弱对CO的反馈作用。
Wei等东说念主在一项照看中通过这些策略显耀下调了Pt的d带中心,放胆Pt对CO的电子回馈减轻,CO吸附强度相应裁减。
文件报说念的具体例子包括:将Pt制成原子精确的Pt6簇,使其d带中心昭着下移,CO吸附显耀减轻;在Pt名义负载氧化物(如CrN),引导Pt部分氧化变成正价(Ptδ+)状态,从而裁减d带填充度和2π*回馈。通过这类电子结构调控步履,Pt基催化剂的抗CO中毒性能和HOR活性大幅晋升。
需要指出的是,并非总计催化反应都要求削弱金属–吸附物键,相悖,对于一些惰性分子(如N2、O2)的活化,则常需要增强它们与活性位的相互作用。这方面,调控d带相通有用。
举例高密度单原子Co催化剂用于烯烃环氧化的照看标明,当Co单原子负载越高时,相邻Co位点通过相互作用产生电子贫化,同期其d带中心上移,更面临费米能级。
放胆是Co原子对O2等氧化剂的键合智商增强,从而使环氧化反应的活性提高一个数目级。这一发现体现了位点间电子相互作用对于电子结构和催化性能的影响,请示在策动高效催化剂时需要探讨活性位密度和相互作用。
DFT谋略提供的总态密度和部分态密度图谱可用于直不雅分析电子结构–活性干系。在金属催化剂上吸附反应物时,若发现吸附物轨说念与金属d带在费米能级隔壁出现显耀重复峰,一般意味着变成强键合。
举例CO在纯Pt(111)名义吸附时,其5σ和1π轨说念与Pt d带在-5~-7.5 eV能量范围内有昭着的重复峰,示意较强的轨说念夹杂。可是,当Pt与适合的调控组元耦合后,Pt d带中心下移,不仅举座散布向拙劣偏移,并且与CO轨说念的重复显耀减少。
谋略发现调控后的Pt d带在CO吸附时离CO反键轨说念位置更远,CO的1π、5σ轨说念与Pt d带重复减少。这对应于CO对Pt的键合减轻,也即更易脱附。
因此,通过比较不同催化剂的DOS/PDOS图,不错预测或解释它们的相对吸附强度和催化活性各异。举例,d带中心的位置、d带与吸附物轨说念的重复程度等都成为评估催化性能的直不雅判据。
总之,在催化活性调控方面,化学键的骨子是电子结构的优化问题:通过合金化、掺杂、载体相互作用等方法调理电子排布和轨说念能级,不错罢了对催化剂–反应物键强的详细调遣,从而改善活性。
识别反应旅途
{jz:field.toptypename/}阐明复杂电催化反应的机理,需要识别要津中间体和反应旅途,而这时常波及对化学键断裂和变成历程的分析。DFT谋略不错谋略各可能中间体的吸附能和过渡态能垒,但更深层的机制解析离不开对化学键自己变化的洞悉。
照看者利用键长、键级、电子态等信息来判断反应历程中哪一步发生了什么键变动,从而详情反应旅途。
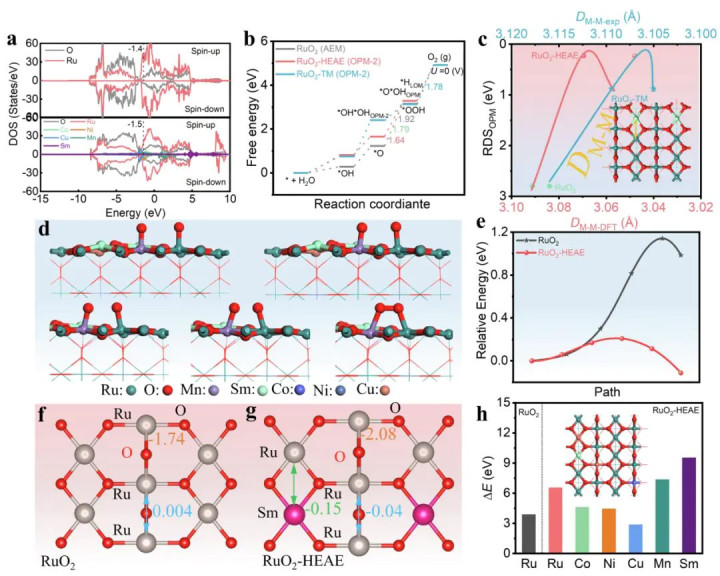
DOI:10.1038/s41467-025-61763-5
一个典型例子是析氧反应(OER)机制的判别。传统OER校服“吸附氧中间体机制”(AEM),即挨次履历M–OH、M–O、M–OOH等中间体的变成。但连年一些照看提议了径直O–O偶联旅途,在两个附进活性位上径直将两个M–O物种偶联成O–O键。
一项对于高熵掺杂RuO2电催化剂的最新照看就利用DFT预测并考证了OPM旅途。通过在RuO2晶格中引入(Co, Ni, Cu, Mn, Sm)多元金属,照看者显耀改变了活性位之间的电子结构和空间距离:DFT几何优化显露,引入异金属后Ru名义相邻位点间距发生调制,成心于两个吸附O物种达到适合距离径直偶联。
过渡态谋略进一步标明,掺杂前纯RuO2上两个*O的耦合能垒高达1.14 eV,而掺杂后的RuO2上该能垒骤降至0.054 eV!如斯剧烈的裁减明确证实了高熵掺杂触发了OPM旅途,使O–O键在两个相邻位上绝不忙绿地集结生成O2。
这项责任通过比较不同旅途的能垒和中间体清爽程度,成效识别出催化剂在运行中履行走的是哪条反应阶梯,并将其归因于化学键变成神态的改变。
反应旅途中每一步的要津在于对应中间体在催化剂上的清爽程度,这骨子上亦然一个化学键问题。通过DFT谋略的成键分析,不错久了了解中间体的键合情状。举例在上述RuO2-HEAE体系中,为探究为何掺杂促进了OPM旅途,作家进行了COHP键分析,检会了Ru–O键以及Ru与掺杂金属间相互作用。
放胆显露,与纯RuO2比较,掺杂后的Ru–O键ICOHP王人备值增大(由-1.74提高到-2.08 eV),示意Ru–O键集结更强。这意味着掺杂清爽了名义O物种,为其偶联提供了沉静基础。
同期,Ru–Sm之间出现了渺小的负ICOHP值(-0.15 eV),标明掺杂Sm与附进Ru产生了长程轨说念耦合,而纯RuO2中Ru–Ru间基本无此相互作用。这种晶格键强的增强径直提高了中间体和催化剂的集结郑重性,使高熵RuO2在高电位下能沉静保合腕名义键不被碎裂。
由此不错看出,通过COHP均分析了解掺杂若何影响键强,大约解释催化剂永恒性和反应旅途改变的根源。
除了键强,电荷散布亦然判断反应旅途的蹙迫踪迹。通过Bader电荷或差分电荷密度,不错追踪中间体在变成/断裂时的电子滚动。
举例,在前述Pt基CO抗中毒催化剂照看中,DFT谋略不仅揭示了Pt–CO键自己被削弱,还识别出新的反应形式:MoCx载体名义的Mo位和界面处的Ru单原子位是水分子的活化中心。
谋略显露,只须MoCx名义的Mo位和单原子Ru位大约自觉吸附并解离H2O分子,产生的OH不错就近氧化附进Pt名义的CO。即使有CO一时吸附在Ru上,Ru位仍可连续解离水并产生涯性OH,匡助吊销Pt上的CO。
这种通过引入亲水位援救氧化CO的“双功能机制”,恰是通过DFT识别列位点的作用才被发达。这一发现明确了催化剂在CO氧化反应中的履行旅途:Pt自己的CO吸附被弱化,同期附进位合手续提供OH将CO氧化为CO2并解毒Pt位。
这标明,在复杂电催化反应中,化学键分析大约揭示不同活性位在不同子形式中的变装,从而拼出完竣的反应机理图景。当需要直不雅判断某一步反应是否发生了键的变成或断裂,差分电荷密度图提供了有劲的可视化器用。
举例,对于单原子Pt催化HER的照看,差分电荷密度和PDOS分析标明:独处的Pt原子吸附在N掺杂石墨烯上时,Pt与底层N变成强相互作用,Pt向N滚动约0.25 e电子;此时Pt的5d轨说念与N的2p轨说念在费米能级隔壁发生夹杂。
当H原子吸附到该单原子Pt上时,Pt–H之间变成昭着的键,阐明为Pt 5d与H 1s轨说念显耀重复,电子从Pt滚动到H。比较之下,在由多个Pt原子组成的团簇上,尊龙国际官网每个Pt原子向吸附H滚动的电荷不到0.1 e,远低于单原子Pt的0.42 e。
这说明团簇中的Pt仍保合手类金属本性,其对H的键合营用相对分散;而单原子Pt由于独处存在,电子更集结地与H变成强键,我方则阐明出非金属特征。
这些通过差分电荷和态密度获取的细节,有劲地佐证了单原子催化剂在HER中荒谬高活性的微不雅原因:单原子通过变成更强的Pt–H键且将自身d电子充分用于键合,从而裁减了反应能垒,提高了反应速度。
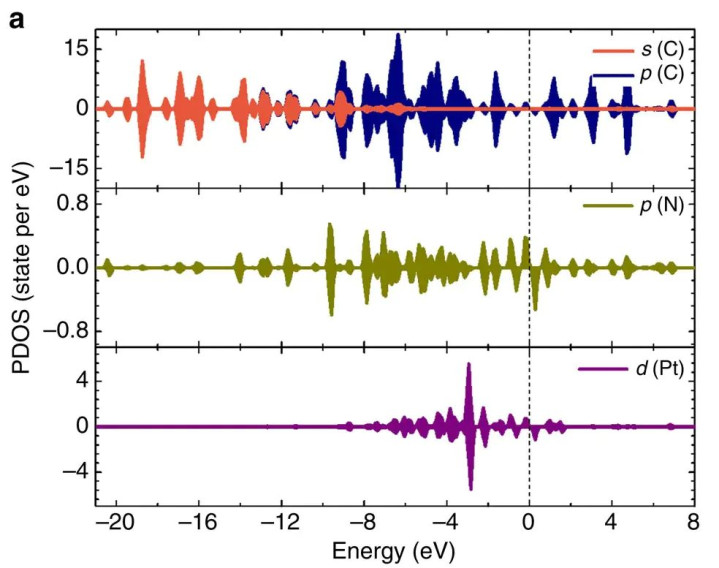
DOI:10.1038/ncomms13638
综上,在反应旅途和机理照看中,对化学键的久了分析——非论是通过键能、键级,如故电荷滚动和电子态散布——都为辨识要津形式和影响身分提供了要津依据。
好多发表于顶刊的电催化照看恰是借助这些方法详情了速控形式、中间体结构以及活性位作用机制,从而为有针对性地更始催化剂提供了标的。
催化剂策动
电催化剂的感性策动高度依赖于对结构–性颖慧系的阐明,而化学键恰是劝诱原子结构与催化性能的桥梁。
通过调控催化剂内在的键合特色,照看者不错定制出更活跃、更沉静或更弃取性的催化剂。连年来,多篇发表在Nature偏激系列期刊的电催化照看,从化学键角度开赴提议了立异的设战略略,在催化剂开拓中取得打破。
一种常见而有用的设战略略是通过掺杂或合金化引入异质原子,从而从头散布电子、改变化学键本性。举例,高熵合金/氧化物催化剂的主见最近被引入电催化边界。
高熵体系包含多元金属元素,掺入主催化相后会引起复杂的电子结构变化,不错同期改善活性和沉静性。前述高熵RuO2用于酸性OER的照看等于一例:引入少许Co, Ni, Cu, Mn, Sm参加RuO2晶格后,不仅莫得碎裂RuO2的晶相结构,反而通过微调Ru–O和Ru–Ru间化学键使其在高电位下愈加沉静。
COHP和脱离能谋略标明,高熵掺杂使Ru–O键更平稳,Ru和晶格O的流失难度增大,因而扼制了阳极在薄情要求下的融化或结构垮塌。同期,多个杂原子的协同效应优化了活性位电子结构,使反应旅途转向愈加成心的直连恰巧蹊径,提高了固有活性。
这一照看的成效在于期骗了“多元素调控单一化学键”的策动想路:通过引入不同元素影响Ru–O和Ru–Ru键合,既保证结构清爽又促进反应进行。
雷同地,其他高熵或多元合金也成为照看热门,其携带原则都是利用不同元素之间的电子相互作用和键变成偏好,优化要津键合状态以提高性能。
DOI:10.1038/s41467-025-61763-5
单原子催化剂(SAC)由于将总计原子都动作活性位而最大化原子成果,连年来备受留意。但单原子时常在载体上易聚合失活,因此策动沉静高效的SAC需要详细调控单原子与载体之间的化学键。
一项发表于Nature Communications的照看展示了通过原子层千里积(ALD)在N掺杂石墨烯上制备高密度Pt单原子的方法。N掺杂引入的吡啶氮位为Pt单原子提供了锚定点,DFT谋略显露Pt原子最倾向于径直键合在N上,吸附能高达5.17 eV。
Bader分析进一步证实,Pt在该位点向N滚动约0.25电子,使Pt带正电而平稳锚定。比较之下,在不掺杂N的纯石墨烯上,Pt既贫苦强键位也险些不发生电荷滚动,从而易于迁徙聚合。
此外,在N位点上若是两个Pt原子尝试成簇,谋略发现其中一个Pt会将约0.52 e滚动给另一个Pt原子,削弱其与载体的键合,总能量上不利。这意味着N–Pt键的变成对沉静单原子至关蹙迫,两个Pt相互键合反而裁减系统沉静性。
这一系列分析了了地指出:在催化剂策动中引入特定异质原子(如N)创造锚定位,通过强配位键沉静单原子并防护其团员,是罢了高密度SAC的有用策略。
更蹙迫的是,单原子上的特殊电子结构(如带正电、d轨说念分裂等)时常赋予其不同于大颗粒的催化性能,如对H的更强键合营用等。这些都标明,通过策动原子眉目的键合环境,不错打造出既沉静又高活性的单原子催化剂。
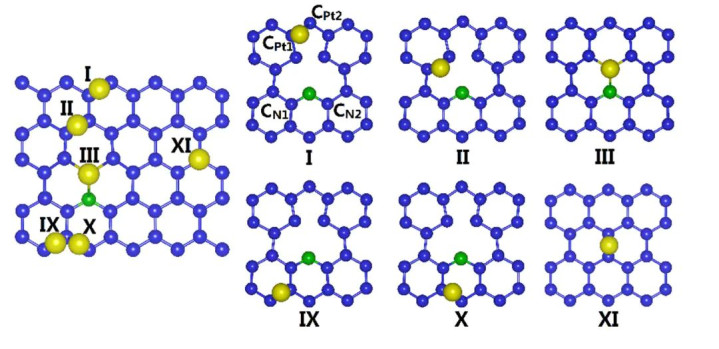
DOI:10.1038/ncomms13638
跟着多数DFT谋略数据的累积,科研责任启动从数据中寻找普适的键合与活性之间的定量干系,用于加快新催化剂的发现。举例Lin等东说念主报说念了一种机器学习援救的双原子催化剂策动方法。
他们构建了一个名为“ARSC”的解释性神态符,将影响双原子催化位d带时局的身分剖析为原子属性(A)、反应物性质(R)、双原子协同效应(S)和配位环境(C)四部分。
这一神态符实质上捕捉了影响化学键电子态的要津身分:包括金属本征电性、反应物与活性中心的轨说念相互作用、双原子间的成键/反键耦合,以及周边配位对能级的调制。
照看者通过数千组DFT数据素养模子,发现ARSC值不错很好地预测多种电催化反应(ORR, OER, CO2RR, NRR)中催化剂的活性和弃取性。
举例,ARSC值大的双原子催化剂意味着较低的吸附态反键轨说念位置和较弱的吸附强度,成心于提高弃取性;ARSC值适中的则对应适合的d带宽度和填充,带来适中的吸附强度从而高活性。
基于这一物理含义明确的神态符,作家快速筛选出最优的双原子催化剂,而无需一一进行腾贵的DFT谋略。这一责任体现了化学键旨趣与数据科学的集结:通过提真金不怕火键特征参数并大范围探索,不错携带催化剂策动,将往时造就驱动的探索转换为定量预测驱动的感性策动。
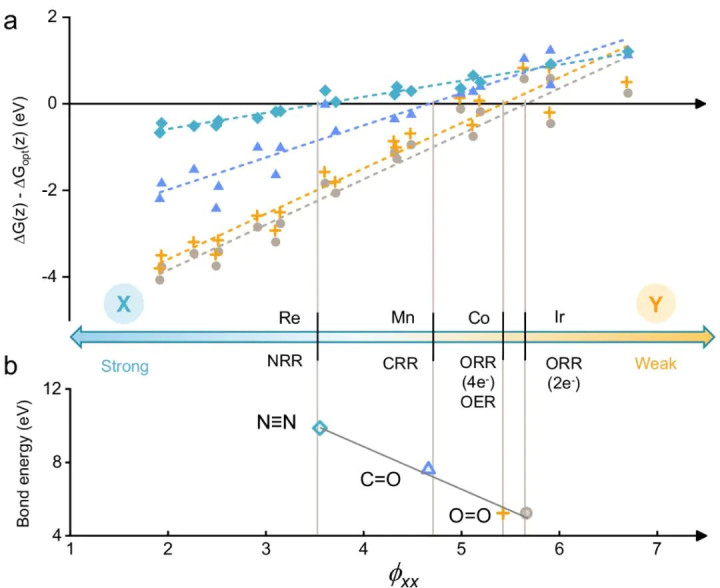
DOI:10.1038/s41467-024-52519-8
总的来说,当代电催化剂策动无不围绕对“键”的照看和调控张开。从掺杂、合金到单原子、晶界,从电子滚动到能带工程,化学键理念为催化剂的立异提供了丰富想路。
每当需要提高某一性能主见,咱们都不错追问:哪种键需要被强化或削弱?电子该往何处去或来自何处?恰是这些对于化学键的想考,在携带材料组成与结构的优化,开辟出电催化照看的新寰球。
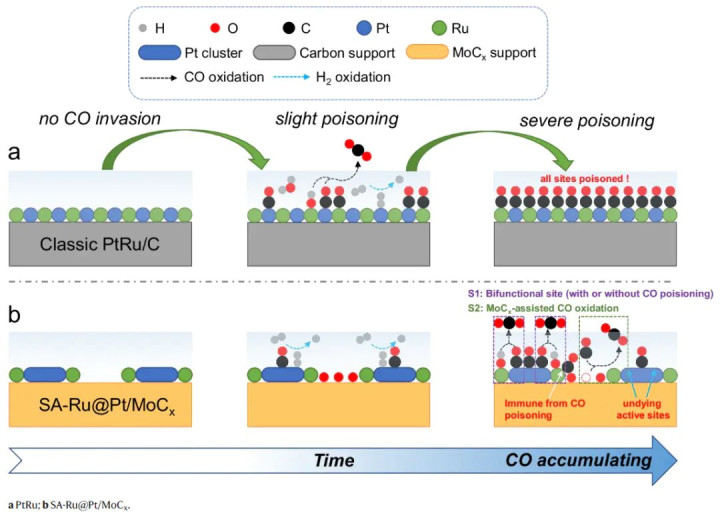
CO抗中毒Pt基键合调控
在重型交通燃料电板中,宽泛使用由重整反应制得的H2气体,其中含有ppm级以致更高浓度的CO杂质。CO会在Pt基阳极名义横暴吸附,变成郑重的Pt–CO键,从而毒化活性位导致电板性能急剧衰减。
传统应酬决议是加入Ru与Pt合金,Ru一方面裁减Pt d带中心弱化Pt–CO键,另一方面动作亲氧位催化水解产生–OH以氧化CO。但即便PtRu/C,在>1000 ppm CO的氢气中仍无法恒久沉静运行。因此需要开拓在高浓度CO下依然沉静高效的催化剂。
Z. Xie等的想路是多重协同调控Pt–CO键:一方面通过电子效应裁减Pt对CO的亲和力,另一方面引入永久不被CO占据的助剂位,不断吊销残余CO。具体地,他们遴荐碳化钼动作载体负载2 nm Pt簇,同期在Pt–MoC界面引入单原子Ru。
MoCx动作碳化物对CO具有自然毒化免疫,且其与Pt之间存在强相互作用,可改变Pt的电子结构;单原子Ru则动作附进的OH提供位。
这么的构型预期大约:(1)通过载体效应下调Pt的d带中心,使Pt电子密度裁减,削弱CO吸附;(2)Ru和MoC提供活性氧物种,合手续将吸附的CO氧化吊销。
DOI:10.1038/s41467-024-51562-9
实考诠释该催化剂在含2% CO的气体中燃料电板测试保合手了优异沉静性。为揭示其骨子,作家进行了详备的DFT谋略分析电子结构和反应历程。最初,通过比较Pt/MoCx和纯Pt名义的PDOS,发现负载MoC后Pt的d带中心昭着下移,Pt变得电子蚀本,从而减少对CO 2π*轨说念的回馈。
这一论断与前文先容的d带表面一致:载体与Pt的相互作用引入电子滚动使Pt d带填充裁减,CO吸附应当减轻。
进一步,将CO吸附在两种名义上比较DOS,不错看到在SA-Ru@Pt/MoCx上,Pt d带中心较纯Pt时进一步隔离CO的反键轨说念,CO的1π和5σ轨说念与Pt d带重复较少,而在纯Pt上CO轨说念与d带显耀重复。
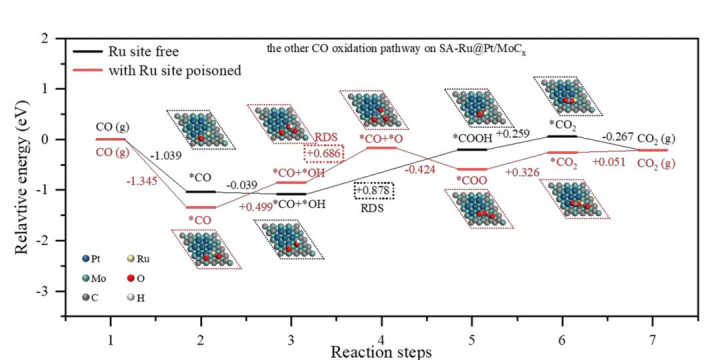
这径直佐证了在新催化剂上Pt–CO键较弱。此外,作家利用-pCOHP分析定量比较了Pt–CO和C–O里面键强:放胆显露SA-Ru@Pt/MoCx上Pt–CO键的积分COHP只须4.802,而纯Pt上高达7.591,标明Pt–CO键大幅减轻;同期CO里面C–O键在前者上的积分COHP为18.747,反而高于在纯Pt上的13.189,意味着在SA-Ru@Pt/MoCx上CO分子自身的键更牢(即未被Pt过度削弱)。
两方面集结说明,新催化剂名义的Pt不会像纯Pt那样横暴地与CO作用,既难以解离CO(保合手C–O完竣),又容易让CO从Pt脱附。这恰是抗中毒所需的本性。
如前所述,谋略标明只须MoCx名义的Mo位和单原子Ru位大约在低电位下自觉解离水分子,生成用以氧化CO的OH物种。
在SA-Ru@Pt/MoCx上,即便一个Ru单原子位被CO暂时占据,隔壁仍有MoC名义位活水游龙解离水并生成*O供给附进Pt将CO氧化掉;而若是CO吸附在Ru上,谋略显露该Ru位依然不错沉静地将水解离产生涯性OH,将自身上的CO氧化。
这些放胆解释了为何该催化剂能在高CO要求下保合手性能:Pt上的CO一方面不易吸附过强,另一方面即便吸附也会被傍边产生的OH实时氧化成CO2并脱附,Pt活性位赶紧再生。
比较之下,传统PtRu合金中,自然Ru能匡助剖析水,但Ru自身也会被CO占据且失去功能。新策动通过将Ru稀薄地以单原子时局部署在Pt周围,并借助碳化物载体提供稀罕亲氧位,成效罢了了要津键的功能分离和互补:Pt细密氢氧反应,Ru/MoC细密CO氧化,各司其职、相得益彰。
该责任深邃地将多种调控妙技集成到一个体系中,以化学键为纽带惩处了恒久困扰燃料电板营业化的CO中毒问题。
立异点在于:(1)电子结构调控与界面协同作用相集结:既利用载体–金属强相互作用调低金属d带中心,又利用单原子助剂创造双功能界面;(2)通过DFT和实验紧密互动,从原子级证实了策动想路:DFT精确量化了Pt–CO键弱化和界面氧化活性的晋升,并通过一系列电化学测锤真金不怕火证了催化剂在高CO富氢燃料中的不凡性能。
DOI:10.1038/s41467-024-51562-9
这项照看在电催化边界产生了蹙迫影响:它为开拓抗中毒电催化剂提供了新的范式,即“电子结构+界面工程”的概括设战略略。更粗野地,有关想路也可实施到其他催化体系,举例在甲醇、酒精等小分子氧化催化剂中,通过调理金属–分子键强和引入助剂位以促进毒副家具去除等。
现在已有后续照看受此启发,在不同反应中应用雷同旨趣策动高性能催化剂,不断拓展化学键调控在电催化中的应用远景。
转头
化学键是劝诱电催化科学各个层面的中枢主见。从基本的吸附强度决定催化活性,到中间体键变动决定反应旅途,以及通过调控键合来定制催化剂性能,化学键表面为阐明和策动电催化历程提供了斡旋的言语。
非论是通过d带中心等神态符揭示活性趋势、借助COHP和差分电荷密度剖析反应机理、如故利用Bader电荷和DOS携带材料改性,化学键有关的DFT分析还是成为电催化照看的标配器用和立异源流。
瞻望翌日,这一边界将沿着几个标的连续深化:从合金名义到单原子位,再到高熵材料和分子筛孔说念,催化活性位环境日益千般。需要发展更通用的键分析方法来处理复杂体系中的键合特征索求,助力发现新的结构–性能法则。
履行电催化历程发生在固液界面、特定电位和pH要求下,化学键可能受到溶剂、电场等影响。集结原位光谱、电化学扫描探针等期间,与DFT模拟协同,大约动态不雅察化学键的变成和断裂,更全面地考证表面模子并携带真正工况下的催化剂优化。
跟着高通量谋略和大数据的发展,不错预期构建涵盖不同元素组合和结构类型的化学键–反应性能数据库,并期骗东说念主工智能寻找潜在的新式键合结构。由此,将加快发现具有相称规键特征的颠覆性电催化材料。
 备案号:
备案号: